
Specification Setting During Chemical API Development

Over the last twenty years or so, the paradigm behind setting specifications has undergone something of a transformation. Much can be attributed to the adoption of risk-based methodologies, such as Quality by Design (QbD), advancements in technology, and harmonization of world regulatory expectations.
The Art of Medicinal Science
Specification setting is – in some measure – an art. While regulatory guidelines delineate what specifications should entail to ensure patient safety (e.g., ICH Q6A specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances), applying specifications throughout pharmaceutical development requires interpretation and judgement applied by various scientific subject matter experts. Developing suitable specifications requires not only an understanding of process and analytical chemistry, but also a grasp of regulatory expectations combined with in-depth knowledge of the realities at commercial-scale versus bench to manage process variability.
What is Specification Setting?
Setting specifications is the practice of establishing conformance criteria for a drug substance, or API, to ensure a safe and effective product. The specifications describe the analytical procedures and limits an API must meet to conform to the specification. According to ICH Q6A:
“A specification is defined as a list of tests, references to analytical procedures, and appropriate acceptance criteria, which are numerical limits, ranges, or other criteria for the tests described. It establishes the set of criteria to which a drug substance or drug product should conform to be considered acceptable for its intended use. “Conformance to specifications” means that the drug substance and / or drug product, when tested according to the listed analytical procedures, will meet the listed acceptance criteria. Specifications are critical quality standards that are proposed and justified by the manufacturer and approved by regulatory authorities as conditions of approval.”
 ICH addresses commercial product specifications and may not directly apply to early stages of development or upstream control points in the process (i.e. raw materials, intermediates, and in-process controls). Development chemists must keep their eyes on ICH limits to keep the end in mind and manage risks during clinical studies. As the relationship between product quality with raw materials and process parameters becomes better understood, specifications are applied to raw materials, isolated intermediates, and various checkpoints throughout the process. These raw material, intermediate and in-process controls ensure a capable process producing product of consistent quality.
ICH addresses commercial product specifications and may not directly apply to early stages of development or upstream control points in the process (i.e. raw materials, intermediates, and in-process controls). Development chemists must keep their eyes on ICH limits to keep the end in mind and manage risks during clinical studies. As the relationship between product quality with raw materials and process parameters becomes better understood, specifications are applied to raw materials, isolated intermediates, and various checkpoints throughout the process. These raw material, intermediate and in-process controls ensure a capable process producing product of consistent quality.
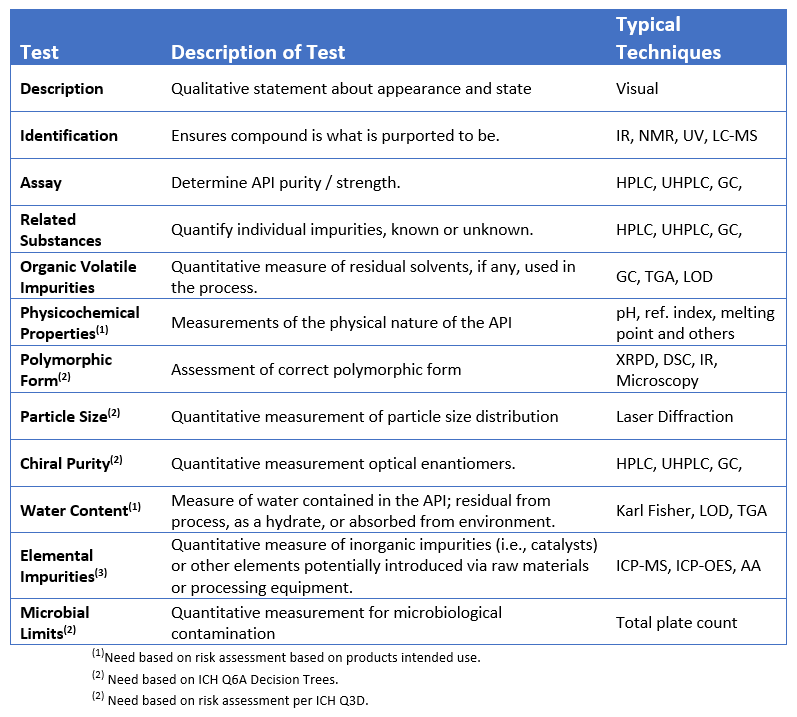
Typical Test Methods
There are a number of common tests used in analytical chemistry when producing small molecules. These may include:
Challenges with Specification Setting
There are several key factors the pharma industry must consider when establishing drug substance specifications.
- Specifications Require Justification
In addition to understanding precisely what the specification is, regulatory agencies want to know why it has been established. Justification for specifications should be based on a range of sources, including accepted criteria already established in existing regulatory guidance. Justification may also be required for as to why a specification is not required for a specific attribute.According to the ICH: “When a specification is first proposed, justification should be presented for each procedure and each acceptance criterion included. The justification should refer to relevant development data, pharmacopoeial standards, test data for drug substances and drug products used in toxicology and clinical studies, and results from accelerated and long-term stability studies, as appropriate.”
- Setting Specifications in Development; Fit-For-Use
Over the course of pharmaceutical development, quality and safety standards are established by the outcomes of preclinical and clinical studies. Setting restrictive specifications too early in development and before process capability is fully understood can present downstream challenges in manufacturing leading to rework and project delays. Consider demonstrating safety using less pure products than expected during future manufacturing campaigns. Early on and when not a risk to patient, it may be appropriate to collect data rather than applying a specification. Later, and as the process is well understood, specifications can be more easily added than removed. To ‘solve for 100% purity’ early in the project may take much more time and money.
- Avoiding the Urge to Overdo it
Just because you can doesn’t make it a good idea. The accuracy and sensitivity of a method can present significant challenges in the lab, even when the process is more than capable. Vendors produce equipment with standards and guidelines in mind. Sampling and sample preparation may require additional environmental controls (i.e. clean rooms). Furthermore, generally accepted validation criteria may no longer apply and may even require a statistician to develop customized strategies and criteria to demonstrate confidence. Examples include increasing the accuracy or lowering the detection limit of a method. Such actions increase cost and stress lab systems which is difficult to justify without benefits to patient.
- Modifying Specifications While in Production
Changing specifications must be treated with the utmost respect. Think of it alike changing blueprints during high-rise construction. While it might be obvious to some, the development team built a process to meet a certain endpoint. Changing the endpoint may require changes to the process and effort required. Just because products met proposed specifications in previous campaigns, does not mean the process has the controls built in to consistently meet that same criteria in the future. To ensure a specification change is met, additional investigative work should be performed and necessary controls added to the process to avoid rolling the dice on a clinical delivery timeline.
- Introducing New Analytical Methods
At any point of the product lifecycle, the need for a new method may arise. This can be due to unexpected results or new regulations. Introducing a test raises the questions: How do I know my process is capable?… And How do you tie it back to the first batches? This can bring anxiety until data cis collected. Frequently, companies will first find representative samples to examine that do not have commercial implications to enable initial development efforts. Next, a bridging study can be performed in which retained drug substance sample are tested using the new method. This yields data that can be used to support regulatory requirements and guide development teams on whether process improvements are required.
Regis’ Approach to Setting Specifications
The pharmaceutical industry has historically approached analytical method development as a static set of tasks. Once specific methodologies were identified and defined, they were largely set in stone. This approach was common, as it resulted in lower costs and avoided any delays needed to qualify a new method (and requalify the drug substance).
The last few decades have seen vast changes in industry approaches to drug development throughout the product lifecycle imparting a mentality of ‘continuous improvement’. Advances in analytical technology have played a key role, as have risk-based approaches such as QbD, DoE, and other statistical tools. FDA’s risk-based approach to process changes allows, an improvement-focused mindset, in which advancements in methods emerge as more understanding of a molecule or its process is acquired. This can result in the avoidance or reduction of non-conformances.
![]()
At Regis, we use a collaborative partnership-based approach to specification setting involving multidisciplinary teams including Process Chemistry, Analytical, Engineering, Quality and the Client / Sponsor. This team strategizes over each critical quality attributes of the drug substance at Control Strategy Meetings. Regis conducts Control Strategy Meetings each aligned with development and scale-up milestones. This formal review examines the chemical process, development outputs, lot history, customer expectations, and product requirements (dose and route of administration) and arrives at an appropriate strategy for the process at each stage of the development process.
The output of these meetings is a living and breathing strategy that clearly describes the necessary effort, including justification, so all the stakeholders, internal and external to Regis, are aligned on what is necessary to deliver on-time, first time, and in full. The collaborative process functions as a feedback loop, resulting in an improved process and better-defined specifications:
Do you want to learn more about specification setting and how Regis can help solve your small molecule API development and commercialization challenges? Contact us today!